
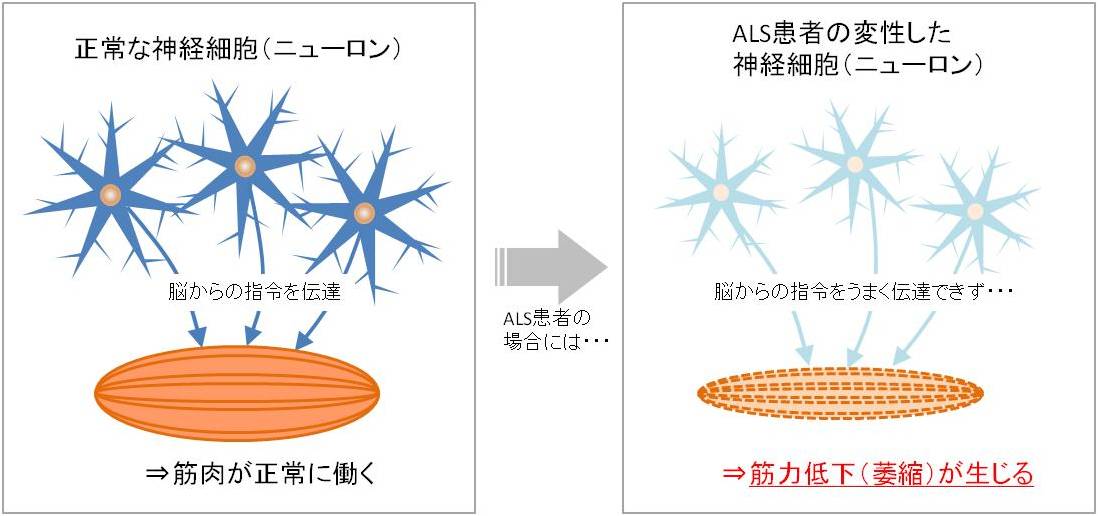
筋萎縮性側索硬化症 (ALS)は、シャルコー病(フランス)またはルー・ゲーリング病(米国)としても知られ、原因不明の後天性神経変性疾患で、主に 脊髄 、脳幹、 脳の運動ニューロンに影響を与えます。 この病気は 神経系 に変性的かつ進行性の影響を及ぼし、不可逆的な運動麻痺を引き起こします。麻痺は徐々に進行し、罹患者の死亡は、話す、動く、飲み込む、さらには呼吸するなど、生命維持に不可欠な能力の喪失の結果です。 ALSの発生率(平均して年間約1/50,000)、有病率(平均約1/20,000)は、西太平洋でより頻繁に発生していると報告されているにもかかわらず、西側諸国では比較的均一です。全体的に男性が若干多いです(男女比は約1.5:1)。 2014 年、保健省は希少疾患を持つ人々のケアを拡大し、ALS を含む希少疾患を持つ人々の包括的なケアに関する国家政策を確立しました。この疾患の臨床プロトコールと治療ガイドラインは 2015 年 11 月に更新されました。

ALS は、最初に現れた臨床症状に従って分類できます。症状が現れ始めた年齢について。 遺伝的多様性 について。進行時間について。そして最も一般的なのは、遺伝様式別(家族性、散発性、グアム)です。多くの遺伝子が ALS に関連していることがすでに報告されています。家族性 ALS は症例の 5% ~ 10% を占め、常染色体優性遺伝、常染色体劣性遺伝、X 連鎖優性遺伝の様式に従って分類できます。このタイプの ALS に属するということは、患者の家族に少なくとも 1 人は罹患者がいることを意味します。症例の大部分は優性型に関連しており、成人に発生します。
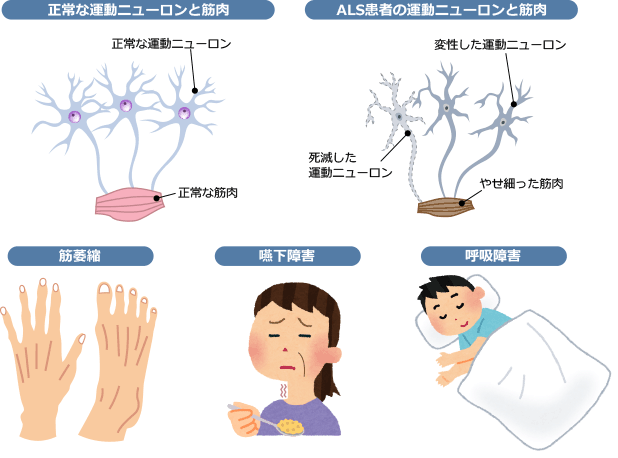
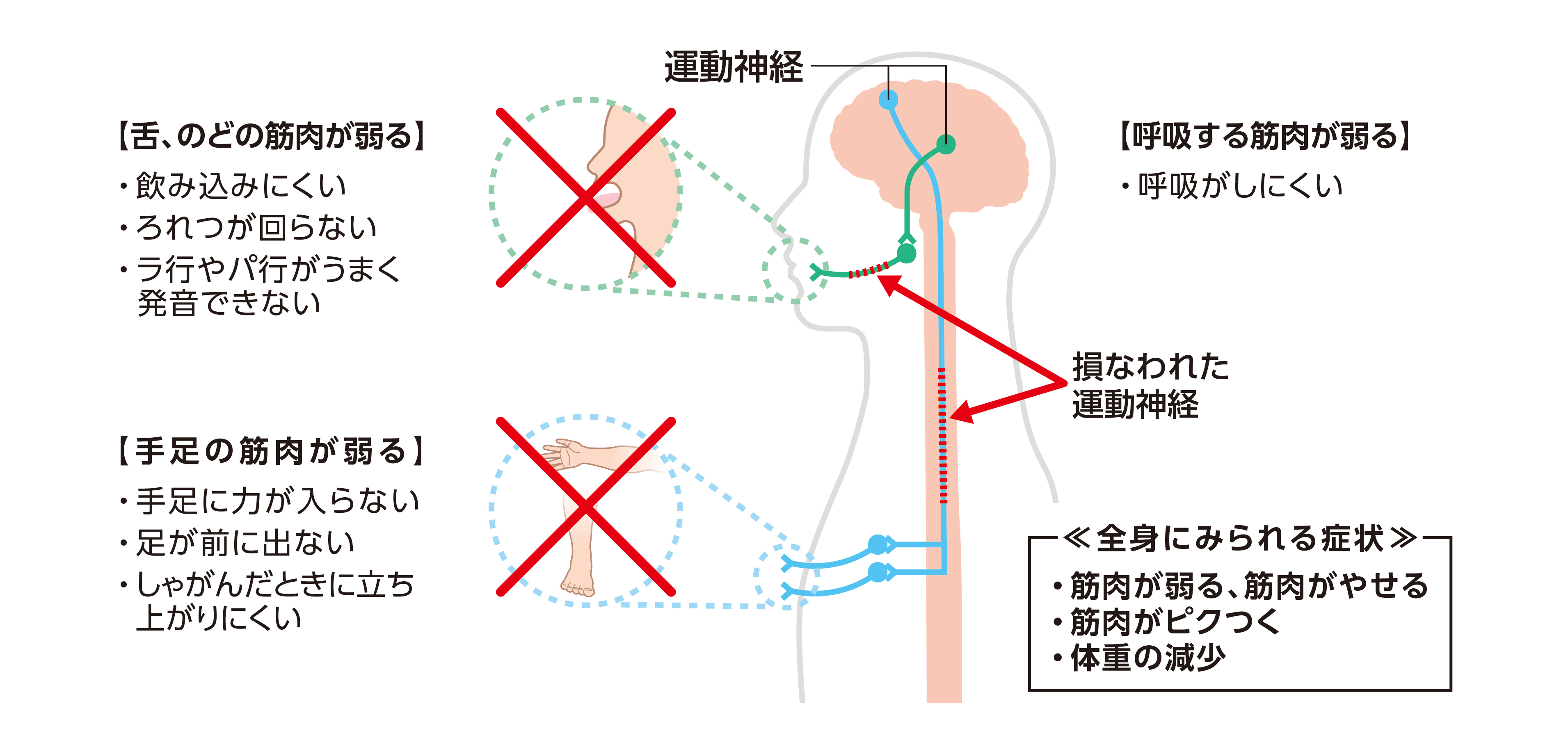
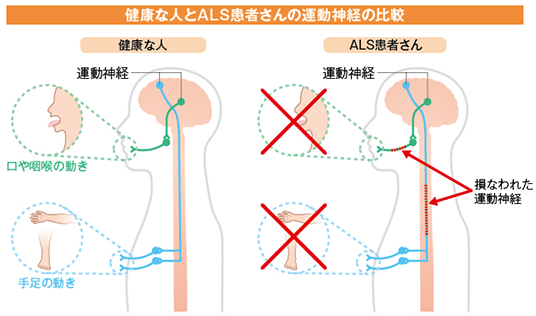
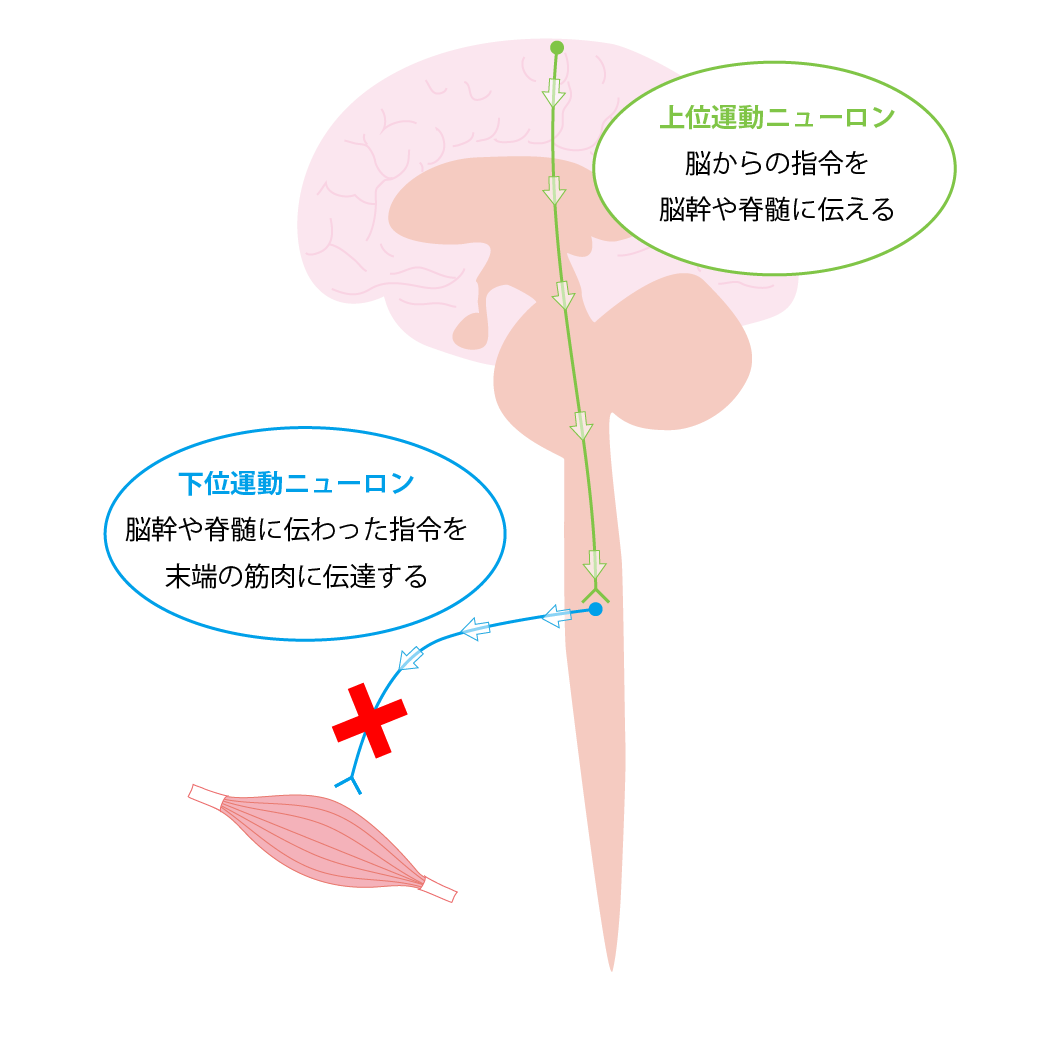
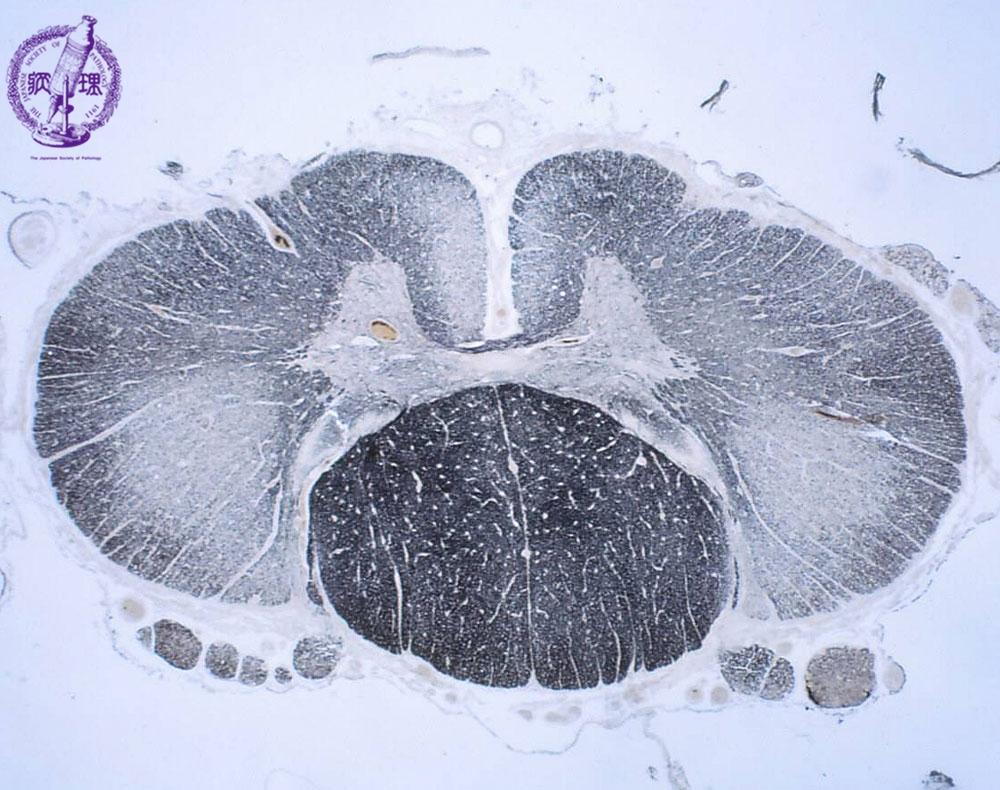
臨床症状、病気の進行パターン、症状発症後の 余命 には多少の違いがありますが、診断は臨床検査によって行われます。 ALS の診断は、病気の経過が長い患者において明らかです。患者の 1 つまたは 2 つの領域 (球、上肢、体幹、または下肢) にのみ局所的な症状がある場合、疾患の早期診断は困難な場合があり、他の罹患領域に兆候が存在するかどうかに依存します。症状の発症から診断が確定するまでの平均時間は約 10 ~ 13 か月です。 ALS の診断は、さまざまな領域における下位運動ニューロンおよび付随する上位運動ニューロンの関与の兆候の存在に基づいて行われます。 ALS の主な兆候と症状は、次の 2 つのグループに分類できます。
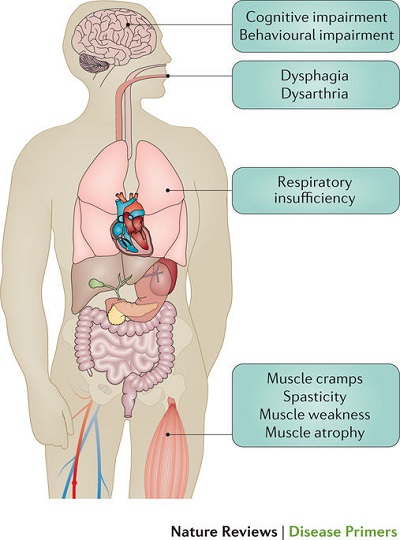
- i) 運動ニューロン変性から直接生じる兆候と症状:脱力感と 萎縮 、線維束性収縮と筋けいれん、 痙縮 、構音障害、嚥下障害、呼吸困難、情緒不安定;
- ii) 主な症状から間接的に生じる徴候および症状:精神障害、睡眠障害、便秘、流汗症、粘液分泌物の粘稠化、慢性低換気の症状および痛み。
初期段階での危険因子と疾患の特定、および専門ケアへの機敏で適切な紹介は、より良い治療結果と症例の予後にとってプライマリケアに不可欠な性格を与えます。現在までのところ、ALS の治療に FDA によって承認されている薬剤はリルゾール 1 つだけであり、症状を軽減し、患者の生活の質を向上させるために、他の薬剤も治療と併用されることがよくあります。 ALSの効果的な治療法を求めて、幹細胞は 生体内 および 試験管内で異なる種類の神経細胞に分化する可能性があるため、数人の研究者が幹細胞を使用しています。 幹細胞 移植はいくつかの動物モデルで効果的であることが示されていますが、修復プロセスの根底にある生物学的経路はまだ解明されていません。 ALS の臨床目標は、運動 ニューロン と非ニューロン細胞 (主にアストログリアまたはミクログリア) の間の相互作用を解明することも目指しています。
参考文献:
1.シェリーDR.筋萎縮性側索硬化症のリハビリテーション:文献レビュー。 ACTA FISIATR 2008; 15(3): 182 – 188.
2. 保健省 – アクセス:02/01/2020 – http://www.saude.gov.br/saude-de-az/ela-esclerose-larate-amiotrofica

3.パルディナJSM。筋萎縮性側索硬化症およびその他の運動ニューロン疾患。出典: Gómes JP、(編集者)。臨床神経学の論文。バルセロナ:医療芸術。 2008.p. 797-826。
4. シラニ V、カルザロッサ C、コバ L、他。筋萎縮性側索硬化症における幹細胞: 運動ニューロンの保護または置換? CNS 神経疾患の薬物標的 2010;9(3):314-2
ギャラリー















")
