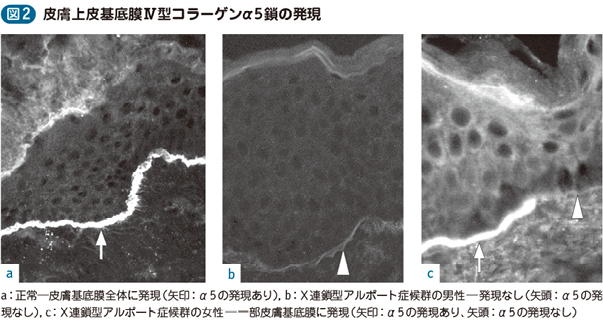
遺伝性腎炎 としても知られる アルポート症候群は 、糸球体疾患の進行性の形態であり、遺伝的特徴があり、一般に水晶体の変化や聴覚機能の喪失を伴います。

この病気は、1927 年にアーサー セシル アルポートによってイギリスの家族で初めて確認されました。
この症候群は、コラーゲン合成を担う COL4A3、COL4A4、および COL4A5 遺伝子に発生する変異に起因します。これらの遺伝子の変化により、IV 型コラーゲン ネットワークの生成が妨げられます。したがって、IV 型 コラーゲン で構成される細胞を支える薄い膜で構成される基底膜は、血液を適切に濾過できず、 タンパク質 が尿中に通過してしまいます。
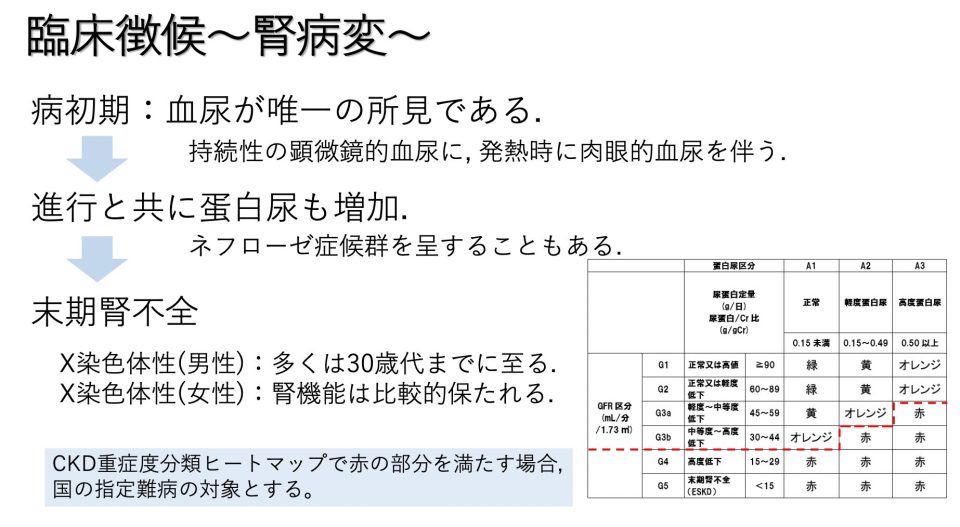
通常、この症状は X 染色体 とともに子孫に伝わります。男性には X 染色体が 1 つしかありませんが、COL4A5 遺伝子の 1 つの変異したコピーだけで重度の Aport 症候群が引き起こされるため、この性別のほとんどの人が発症するという事実が説明されています。腎不全。 X 染色体のコピーを 2 つ持つ女性では、COL4A5 遺伝子のコピーの 1 つだけに変異がある場合、血尿(尿中の血液)の出現のみが引き起こされ、腎不全は発症しません。この突然変異は X 染色体に関連しているため、父親がこの症候群を子孫に伝えることはありません。
この症候群が遺伝する別の方法もあります。これは常染色体劣性型であり、第 2 染色体上に存在する COL4A3 遺伝子または COL4A4 遺伝子の 2 つのコピーが突然変異を起こした場合に発生します。場合によっては、常染色体劣性疾患を持つ子供の親は、常染色体劣性疾患の影響を受けていないものの、変化した遺伝子のコピーを持っていることがあります。

前述したように、この症候群は、視覚機能の変化に加えて、腎臓および聴覚機能の進行性の喪失を特徴とします。その他の症状としては、高血圧、目の周囲、足首や足の浮腫が挙げられます。
この診断は、腎不全、難聴、視力喪失の家族歴によって疑われます。ただし、症例の約 15% にはこれらの変化の家族歴がないため、腎生検が必要になります。
アルポート症候群は継続的なモニタリングによって治療されます。水分摂取量の制限や高血圧の管理に加えて、食生活の変更も推奨されます。
必要な措置を講じないと、この症候群は慢性腎不全に進行し、透析または腎移植が必要になる可能性があります。

ギャラリー